Richard Dawes

Richard Dawes
Program Lead for Chemical Theory, Models, and Computational Methods (CTMC), NSF, Division of Chemistry
Richard Dawes received his Ph.D. from the University of Manitoba in 2005 before joining the group of Prof. Tucker Carrington Jr. as a postdoc at the Université de Montréal. In 2006, he worked with Prof. Donald L. Thompson on fitting PESs before joining Dr. Ahren W. Jasper at the Combustion Research Facility in 2009. He became an Assistant Professor at the Missouri University of Science and Technology in 2010 and was granted an early career award by the U.S. Department of Energy in 2013. His research focuses on multistate multireference quantum chemistry and potential energy surfaces.
Our group is interested in the spectroscopy and dynamics of small molecules relevant to combustion, atmospheric and interstellar chemistry. We are developing methods to construct global potential energy surfaces. In many cases multiple coupled surfaces play an important role in the dynamics.
Calculations of molecular dynamics can be very sensitive to the topography of the surfaces especially at low-temperature. We are exploring high-accuracy multireference electronic structure methods for these applications.
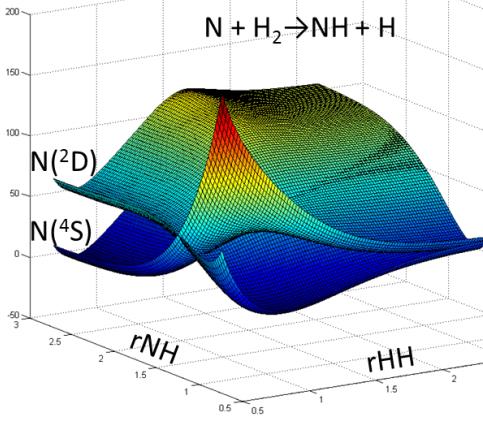
Relevance of Potential Energy Surfaces
Computer simulation plays an increasingly important role in chemical research. We seek to understand the properties and behavior of chemicals and materials in diverse environments such as; hydrocarbon combustion in automobile engines, the dynamics of atmospheric species such as ozone, and the chemistry of interstellar space and other planetary atmospheres. Quantum dynamical simulations can make predictions and provide insight that combined with experiments help complete our understanding of these important systems. The theoretical framework within which we describe the interactions of atoms and molecules is the potential energy surface (PES).
The molecular PES is central to how chemists think about the structure and dynamics of molecular systems, ranging from equilibrium structures at minima to product-channel asymptotes, connected by paths across landscapes and over energetic barriers. The PES describes the energy of the system as a function of its geometry. The energy function once determined allows one to predict stable structures and dynamical behavior. Interactions with radiation such as laser spectroscopy experiments or photochemistry occurring in nature can also be simulated using PESs. Much of our work involves piecing together thousands of individual calculations at different geometries into continuous surfaces. We are working on algorithms that combine hundreds or thousands of individual processors on high-performance computing clusters to complete these tasks in an automated fashion.